

APHA is building increasingly more sophisticated tools to integrate full genome sequences with epidemiological information enabling visualisation of how disease is spreading. In this two-minute read by APHA's Richard Ellis, Head of Genome Analysis and Molecular Biology Discipline Champion, learn about how APHA is using this technology in practice.
When I joined APHA in 2008, I recall saying in my interview: “my mission is to sequence everything that comes in through the front door”, (essentially collect detailed genetic information about the bugs causing disease), to better understand the biology of bacteria and viruses causing disease, and how they are transmitted.
Since then, there has been a revolution in the technology used to generate DNA sequences. The speed of DNA sequencing has increased by several orders of magnitude and the cost per sample has decreased by an even greater margin.
In 2003, APHA was a partner in sequencing the entire genome of a single Mycobacterium bovis sample (the causative agent of bovine tuberculosis (bTB)). This took several years and cost thousands of pounds, whereas today we can sequence 100 samples in two days for less than £100 each.
Over the past 10 years, APHA has invested in this technology and developed robust procedures to generate ‘High-Throughput Sequence’ (HTS) data, meaning that we can generate full genome sequences for a variety of pathogens in less than 48 hours. We achieve this by breaking the genetic material (nucleic acid) into short fragments of a few hundred bases each, and then determine the DNA sequence of millions of these fragments in parallel.
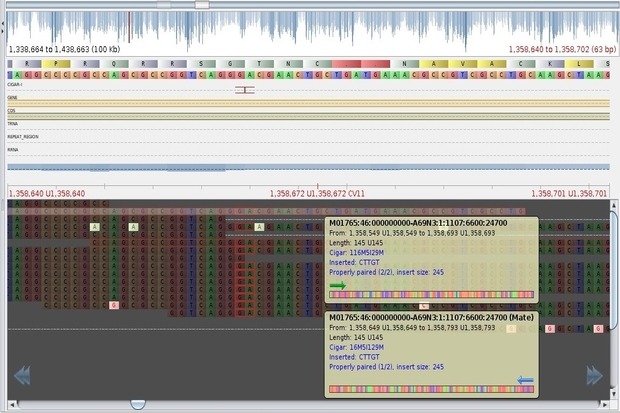
Whilst it is now relatively routine and quick to generate the raw data, the process to stitch the data back together into a consecutive run representing a complete genome (over 4 million bases in the case of M. bovis) is complex and computationally demanding. To meet these demands, APHA has implemented a new IT infrastructure utilising cloud technologies (Scientific Computing Environment, (SCE)).
APHA’s ambition is to utilise the additional information that full genome sequences provide in the understanding and epidemiology of disease outbreaks. We are using bTB as the proving ground for introducing this technology. This disease is endemic across much of England and Wales, and utilisation of whole genome sequencing (WGS) is an important part of governments’ eradication strategy (as outlined in Government Response to bovine TB strategy review - England).
From April this year, all appropriate M. bovis samples have undergone WGS (replacing the previous genotyping) to assign them to one of thirty distinct groups, each with a known geographic range in Great Britain.
Given that the results of genome analysis can lead to decisions about disease control, it is vitally important that we have confidence in the results. Therefore, each separate stage in the process required validation; ensuring that multiple testing of the same sample always leads to the same result, regardless of operator, and that it can be corroborated by other testing methods.
APHA has considerable experience in validating laboratory processes, but it was breaking new ground to attempt this for the computational and analytical processes needed for WGS (bioinformatics) – even external accreditation bodies had not addressed this before. However, we were successful and now have a fully validated end to end process which is externally accredited to ISO 17025 for the ‘Assignment of sequence clade using whole genome sequencing (WGS) of Mycobacterium bovis isolates’. This was an important milestone for our use of WGS and bioinformatic processes and APHA aims to attain this accreditation for whole genome sequencing of other pathogens (e.g. Salmonella and Influenza virus) in the near future.
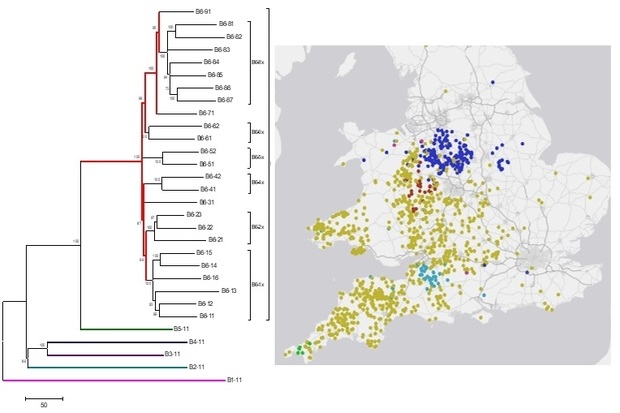
APHA is now building increasingly more sophisticated tools to integrate genome data with epidemiological information, such as location and animal movement history, to enable visualisation of how disease is spreading, identifying key infection pathways and thus potential intervention points. So, whilst we are still not “sequencing everything that comes in through the front door”, we are finally starting to realise the initial vision. This has been a (very) long time in the making and far more complex than I originally envisioned but would not have been possible without a great team at APHA with a wide range of specialisms.
Recent Comments